|
% h4 f: Z8 X+ {- ^& K6 J! l& j" _
Alzforum是一家创立于1996年的新闻和信息资源网站,致力于帮助研究人员加速发现和推进阿尔茨海默病及相关疾病诊疗手段的开发。
9 C3 ^% _4 ~6 P6 S, D
' c4 C/ o- f# L6 W1 H9 _
在2015年12月11日至13日于奥兰多召开的第26届ALS/MND国际研讨会上,科学家、神经学家和ALS患者群体很兴奋地听到关于治疗运动神经元病的临床进展。与会者思量着脊髓性肌萎缩(spinal muscular atrophy,SMA)基因疗法试验初步但积极的结果——可能打开ALS类似基因疗法的大门。他们听到了已经完成和正在进行的依达拉奉(edaravone)试验新闻,该药物可能延缓某些ALS患者的病情发展。他们还听到了缓解语言和吞咽问题的疗法信息。还有基础科学方面的进展,更好地理解致病蛋白质如何在细胞间散播蔓延,同时研究人员介绍了解释一些遗传性ALS风险的新基因变异。 5 X' v* F4 ?& P
* x& s9 T0 D; W. f) B7 z
2015年12月19日——超过800位研究人员、临床医师和ALS患者于2015年12月11日至13日聚集在奥兰多,召开第26届ALS/MND国际研讨会。与会者感受到一种紧迫而有希望的感觉——紧迫是因为“时间不站在我们这一边”英国运动神经元病协会(Motor Neurone Disease Association in the United Kingdom,此次会议组织者)首席执行官Sally Light说道。她指出,仅在英国,每天就有6人因ALS离世。不过,得益于会议上对进展的讨论,包括新颖的治疗方法、识别新的疾病基因和对致病蛋白质如何在细胞间传播越来越充分的理解,与会者感受到研究领域所取得的进展。“我比10年前乐观得多”项目委员会主席、英国牛津大学Kevin Talbot评论道。
`) K+ `! z: [$ t' g
当全国儿童医院(Nationwide Children’s Hospital)Brian Kaspar介绍一项临床试验早期结果时,乐观态度在基因疗法讨论会上表现的最为明显。他治疗了婴儿最严重的运动神经元疾病——脊髓性肌萎缩(spinal muscular atrophy,SMA),罹患该病的婴儿一般会在出生两年内瘫痪和死亡。然而,Kaspar开始于2014年春天的试验中治疗的所有婴儿都保持生存和一定的活动能力(wriggling)。他播放了一名女婴的视频,治疗后6个月,能够不需要支撑地坐直,这是未经治疗的SMA婴儿从未实现的。 “这些是早期的1/2期临床试验”Kaspar警告说,“我们绝不是宣称取得了胜利,但它是令人兴奋的。” e2 L& K+ x7 r3 ]) R' E# i" C7 K
“我认为Kaspar介绍的初步1期数据看起来非常令人鼓舞”冷泉港实验室(Cold Spring Harbor Laboratory)Adrian Krainer评论道,Adrian Krainer没有参与这项工作。“随后需要更大的(安慰剂)对照试验来确定是否存在统计上显著的改善,但初步数据显示没有安全问题,这是很让人高兴的。”在同一场讨论会上,Krainer介绍了一种SMA的反义寡核苷酸疗法,参试者也从正在进行的试验中获益。在另一场单独的讨论会上,Cedars-Sinai医学中心Clive Svendsen描述来他将基因疗法与干细胞(另一种有前景的现代技术)结合起来治疗ALS的工作。他的试验将在2016年初开始。 ( z8 U/ ~0 V. K/ W3 M; X
SMA的基因疗法 # I% B9 K$ }2 w- I
SMA是一种常染色体隐性遗传疾病,由SMN1基因(编码运动神经元生存蛋白)中的错义或缺失突变所致。SMN加工mRNAs,由于未知原因,在包含两个坏的SMN1基因副本时,运动神经元会特别脆弱。一旦运动神经元死亡,失去对肌肉的刺激,肌肉会无力萎缩。人类拥有第二个SMN基因,SMN2,但来自SMN1编码的核苷酸单个差异,会主要导致缺乏外显子7(exon 7)的另一种拼接形式。这种较短的同种型(isoform)不稳定,会快速退化分解。尽管如此,少量SMN2产生的完全长度的SMN蛋白质就足以让携带2个SMN1突变副本的人们生存。事实上,疾病的严重程度基于SMN2表达的多少。有的患者携带的是最温和的SMN1突变版本,IV型,直到30多岁才会表现出症状,但在最严重和最常见的I型中(几乎没有备份的SMN2表达),婴儿期就会被该病摧残。 8 S5 V/ S1 B, h. U) P* N- n
递送工具,科学家们使用腺相关病毒携带基因进入患者。 3 q6 n/ e& z4 t7 R: B) a6 A+ D I6 A% \
在临床试验中,Kaspar和同事们使用无害的腺相关病毒(adeno-associated virus)AAV9来向运动神经元递送一种原始SMN1基因。不同的AAV类型拥有不同的衣壳结构,针对不同的细胞类型。AAV9能够穿过血脑屏障,结合到拥有半乳糖附加的细胞蛋白,将其吸引到运动神经元和星形胶质细胞。虽然在成年小鼠中,AAV9优先转换星形胶质细胞,但在更大的动物中,它也会以运动神经元和少突胶质细胞为靶点,Kaspar说道。
" s9 J7 k# E+ a4 G
一旦找到其靶点细胞,病毒会将其遗传物质塞入细胞核,保留为染色体外的少量DNA。这些DNA会持续存留,所以理论上一次治疗就应该能够提供一生的SMN蛋白。在一种严重SMA的小鼠模型中,治疗保留了小鼠在27周生存期中的活动性,而未经治疗的小鼠生存期仅两周左右。
+ r, t! g8 |& t _+ u
Kaspar的治疗获得基因疗法公司AveXis Inc.的许可,该公司与全国儿童医院就试验(trial)建立了合作。这项研究将招募18名9个月大或更小的婴儿。所有参试者必须是由于两个染色体SMN1基因突变的SMA患儿,症状开始于6个月大之前,肌张力低,运动技能延迟。他们将获得一次性的治疗,静脉输注携带SMN1基因的AAV9,大约一个小时。所有的参试者都接受治疗,不设安慰剂组。不过,这项研究将测试3个不同剂量,从低剂量开始,更后期招募的患儿会接受更高剂量的治疗。研究人员怀疑高剂量可能效果更好,也许是通过转换更多的运动神经元,或可能是通过增加SMN1在单个细胞或感染的神经胶质细胞以及运动神经元中的表达,研究合作者、俄亥俄州立大学Kevin Foust说道。 : A4 s7 Y" H7 M; { H
在最低剂量,3名婴儿获得67万亿病毒颗粒/公斤体重的剂量治疗。科学家们获得FDA批准在9名婴儿中尝试200万亿/公斤体重的剂量,以及在其余婴儿中尝试最高滴定的330万亿/公斤体重剂量。研究者将主要寻找治疗的任何毒性作用,还将评估婴儿离世的年龄或需要呼吸机的时间。他们会将结果与严重SMA的标准发展状况进行比较。SMA研究往往依赖于将死亡或需要呼吸机支持作为作为结合端点。通常,10.5个月大的时候,患儿中的一半会死亡或需要呼吸机,20个月大的时候,92%会死亡或需要呼吸机。此外,试验会通过标准得分来评估患儿运动状况,比如在坐姿时抓握测试人员的手。该试验预计将在2017年6月完成。 9 x5 a0 f! m$ k; |( |8 D; }
Kaspar在会议上回顾了目前为止的数据。3名患儿获得最低剂量的病毒治疗,7名获得中间剂量治疗。研究人员目前还没有尝试到最高剂量。婴儿能够耐受清醒状态的输注,没有问题,Kaspar说道。从2014年4月招募首名婴儿到9月30日的最后数据记录时点,Kaspar说,研究中没有婴儿死亡或需要呼吸机。他们的活动能力也比治疗之前更好,中间剂量治疗的患儿所获得的益处超过低剂量治疗的患儿。虽然严重SMA的患儿从来都无法自己坐起来,但Kaspar视频中的小女孩做到了。试验将继续招募8名患儿,其中一些会接受最高剂量治疗。 & M/ b! u1 n# Q# V8 B/ e4 e
SMN备份疗法
, L2 x' _$ I1 g1 w8 Q
该部分主要介绍针对SMA的反义寡核苷酸(antisense oligonucleotides, ASOs)疗法及试验。详细内容省略。
2 ~* Z5 F/ n0 Q# e
会议上的研究人员对治疗SMA的进展感到兴奋,并表示它们带来了治疗ALS的希望。“SMA是这方面的先驱者” Talbot评论道,“表明遗传疾病,在某种意义上,是容易处理的。”类似的对家族型ALS的治疗已经在进行临床前和早期临床测试。 " [) O" c. B1 a! B# A8 Q
ALS紧随其后? ( @7 o( ?% Z3 L7 Z
Kaspar和同事们自己在开发关闭突变SOD1基因表达的疗法方面取得了进展,该基因的突变会导致10%左右的家族型ALS。他使用AAV9来将沉默SOD1的短发夹RNAs(short hairpin RNAs,shRNAs)递送到ALS转基因小鼠模型的脊髓中。携带人类SOD1-G93A的小鼠通常在19周左右死亡,一次性治疗改善生存期长达7周。在表达SOD1-G37R的病情较温和的模型中,治疗将13个月的平均生存期扩展了3个月。
6 J* x4 L. ^! }1 R
完美转染,科学家们使用AAV9向小鼠脊髓递送SOD1反义寡核苷酸和绿色荧光蛋白(棕色)。
0 v, b, R* Q5 a6 F
AAV9病毒包括一种绿色荧光蛋白(green fluorescent protein,GFP)基因来标记受感染的细胞,这使其不适合用于人类。Kaspar及其合作者们正在测试一种不包括GFP的新版本。在严重型小鼠模型中,一天大的时候单次治疗,使一些小鼠生存期超过6个月。“G93A小鼠生存期活过200天,这确实是对生存曲线的改变”Kaspar说道。如果治疗的安全性在小鼠和猕猴中被证明,他希望推进到临床研究。 6 K' k' ? q$ [0 _! n! K0 v
Talbot指出,动物模型中SOD1表达下降,但没有降至零,研究人员还需要进一步的工作来使这种方法变得完美。“我认为,在神经系统衰老的过程中,少量的错误折叠SOD1就可能触发疾病”他说道。Foust表示,治疗可能无法完全消除疾病,但随时间推移可能减少SOD1积聚并延缓或停止其病程。 ; o8 q$ a& ^$ w2 x
在会议的闭幕讨论会上,Svendsen解释他如何将基因和干细胞疗法相结合。他指出,虽然可以从诱导多能细胞创建运动神经元并移植到脊髓,但这些细胞通常无法融入神经肌肉网络或连接到肌肉。此外,它们可能会受到同样的变性退化影响。因此,Svendsen选择移植星形胶质细胞,这些细胞源于干细胞并经过基因工程(处理)而释放胶质细胞源性神经营养因子(glial-derived neurotrphic factor,GDNF)。GDNF能够促进运动神经元和皮质脊髓神经元生存,但不容易递送到大脑,Svendsen说道。所移植的细胞给它提供了“坐骑”。
( `9 W/ c, V) c; H
在表达SOD1-G93A的大鼠中,所移植的星形胶质细胞存活长达11周并释放GDNF。运动神经元存活时间更长,虽然大鼠依然发展出瘫痪,但负责呼吸的神经比对照组工作得更好。
0 n4 s2 O8 S+ j
Svendsen计划招募18位ALS患者参与1/2期试验。试验将从较低位脊髓的移植开始,但Svendsen怀疑,沿着脊髓的多次植入可能是必需的。Svendsen表示,由于这些大鼠是因为SOD1转基因的多个副本而患病的,因此基于大鼠研究来预测在人类患者中的结果是很困难的。移植可能仅仅是改善呼吸,或者也可能同时减少瘫痪,他猜测。 0 @4 v" a5 a7 P Z) Y+ v
神经外科医生会选择更容易避开血管的一侧脊髓进行移植,Svendsen说道。由于干细胞不会从一侧脊髓跨越到另一侧,因为未植入的一侧脊髓将作为对照。只有外科医生知道哪一侧接受了星形胶质细胞植入,研究中的其他人员都不清楚。ALS症状通常在双腿的发展速度相似,因此研究人员希望治疗能够延缓一侧的病情发展,并使用一个椅子状的设备来测量肌肉力量,监控症状的发展状况。“如果有效果,我们应该能够探测到”Svendsen说道。如果科学家们确实检测益处,他们将为参试者的另一侧脊髓提供移植。他预计这项为期2年的试验将于2016年3月招募首位参试者。 7 p; f1 `5 l. `0 u- g. w
# D7 i0 X6 p: U! u! a0 v( p
$ d% F% Q: P) s7 { n | 




 发表于 2016-1-3 01:46:37
发表于 2016-1-3 01:46:37



 发表于 2016-1-3 02:23:40
发表于 2016-1-3 02:23:40


 发表于 2016-1-3 09:46:40
发表于 2016-1-3 09:46:40
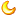

 辛苦了。
辛苦了。{kind=link}